Ovarian Reserve: 3D Instance Segmentation of Oocytes
About this tutorial
This tutorial explains how to use BiaPy for 3D instance segmentation of oocytes in whole-mount mouse ovaries, based on “3D Mapping of Intact Ovaries Reveals the Aging Dynamics of the Ovarian Reserve” (bioRxiv, 2025) [2].
The goal is to make this workflow accessible to all BiaPy users (GUI, notebook, Galaxy, Docker, CLI, or API), even if this is your first time working with 3D instance segmentation.
If you still need to install BiaPy, follow the installation guide and choose your preferred option (GUI, Docker, CLI, notebook, etc.) before continuing with the workflow below.

Left: raw DDX4 image stack. Right: corresponding instance-label stack. |
Note
A pretrained model is now available for download. You can download it from this SharePoint link. This allows you to directly apply the model to your own ovary images without training from scratch. Alternatively, you can train your own model using the provided training dataset and YAML file (see below).
Paper overview
Our publication [2] presents a pipeline to map the entire ovarian reserve in 3D by imaging intact mouse ovaries with light-sheet fluorescence microscopy (SPIM) and segmenting every individual oocyte with a deep learning model. The key steps of the pipeline are:
Whole-ovary SPIM imaging: intact ovaries are cleared and imaged at single-cell resolution across the full organ, yielding large 3D fluorescence volumes (DDX4 channel marking oocyte cytoplasm).
3D instance segmentation with BiaPy: a 3D residual U-Net (ResU-Net) is trained on manually curated oocyte labels using the F + C + Dc channel representation (Binary mask + Contour + Distance to the center), followed by marker-controlled watershed to recover individual instances.
Age-resolved quantification: segmented oocyte counts and spatial distributions are compared across seven age groups (5–60 weeks), revealing the dynamics of ovarian reserve decline.
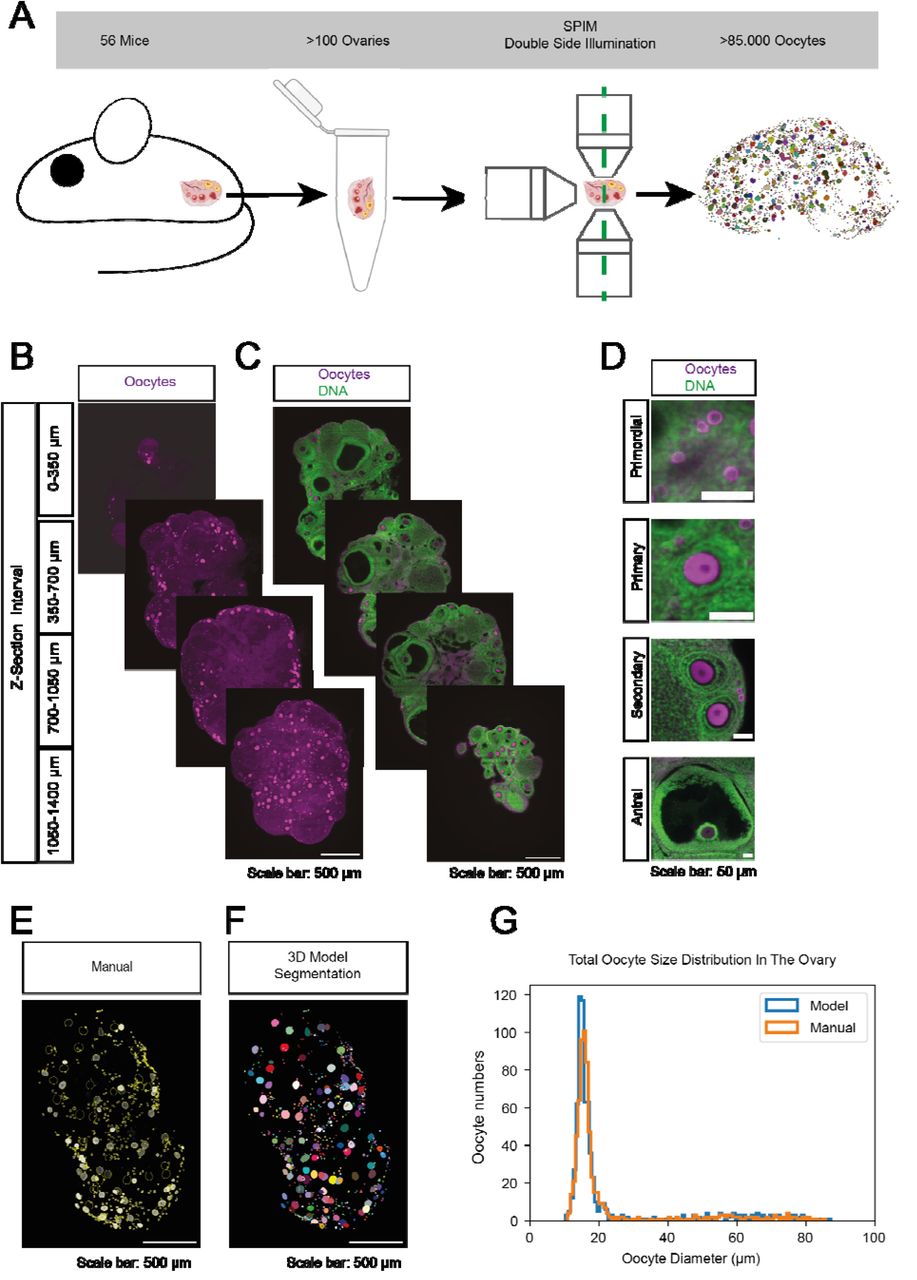
Paper Figure 1 from [2]: SPIM whole-ovary imaging and model-based oocyte segmentation workflow. |
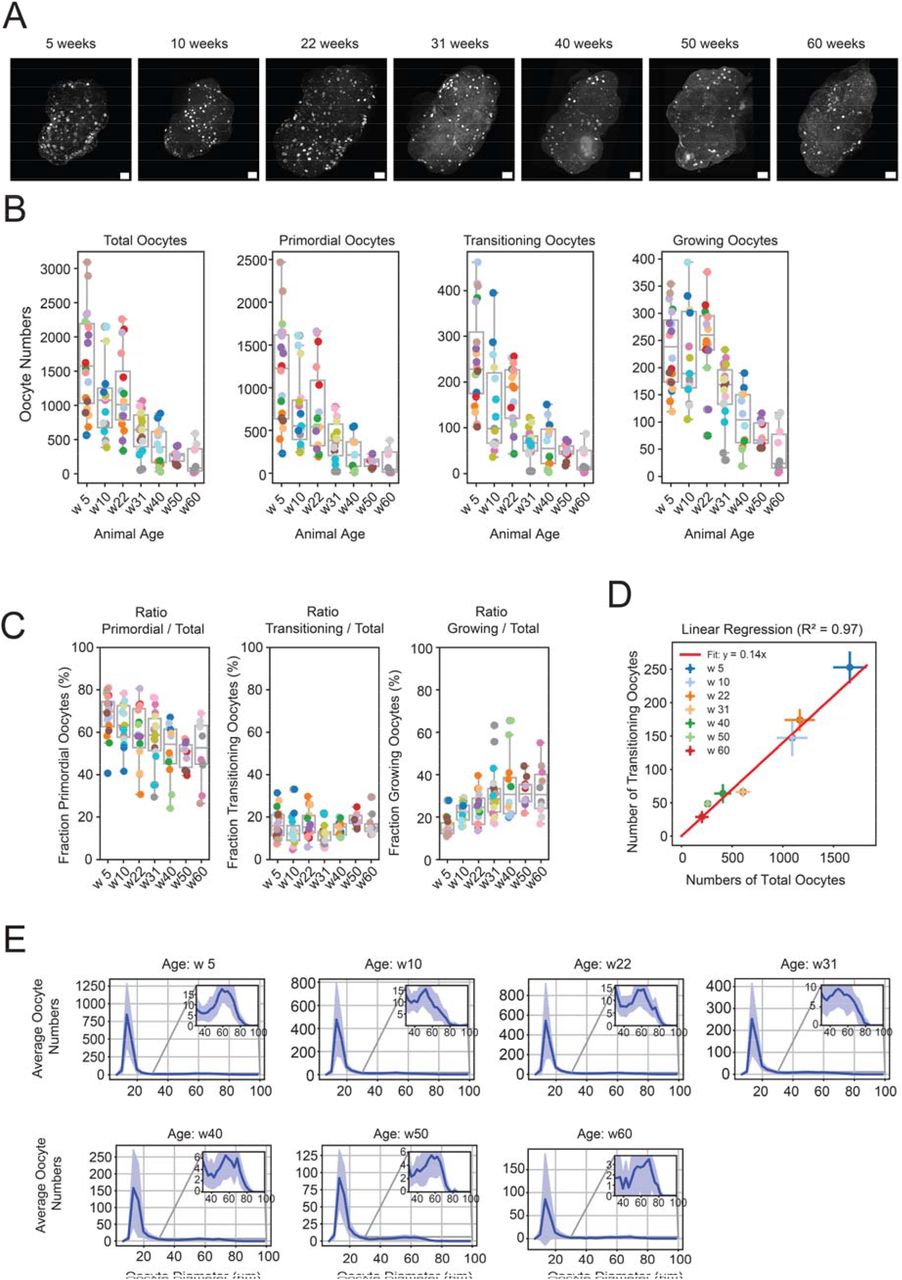
Paper Figure 2 from [2]: age-resolved ovarian reserve quantification enabled by 3D oocyte segmentation. |
Data preparation
This tutorial uses two datasets. Note that they represent a small but representative subset of the full data described in [2] — the complete study involved many more labeled slices and full-ovary volumes. These subsets have been selected so that both training and inference (applying the model to new images to produce segmentations) can be completed in a reasonable amount of time on a standard GPU workstation.
The datasets are presented in the natural order of the workflow: first the training data (used to learn the model), then the test data (used to evaluate it).
Training dataset (sample):
oocyte_training.zip(240.9 MB) containing a curated set of paired 3D raw and label image volumes split into training and validation subsets, sufficient to train or fine-tune the segmentation model: OneDrive link.Once unzipped, you should find the following directory tree:
oocyte_training/ ├── train/ │ ├── raw/ │ │ ├── 10W_100330_frame70.tif │ │ ├── 10W_105114_1.tif │ │ ├── ... │ │ └── 5W_150806_frame54.tif │ └── label/ │ ├── 10W_100330_frame70.tif │ ├── 10W_105114_1.tif │ ├── ... │ └── 5W_150806_frame54.tif └── val/ ├── raw/ │ ├── 10W_100330_fram195.tif │ ├── ... │ └── 5W_130858_frame89.tif └── label/ ├── 10W_100330_fram195.tif ├── ... └── 5W_130858_frame89.tifTest dataset (Zenodo):
raw_ovary.rar(13.0 GB) containing seven full 3D ovary volumes covering a range of ages (5, 10, 22, 31, 40, 50, and 60 weeks) in TIFF format. These are a subset of the ovaries analyzed in the paper and serve as the held-out test set: Zenodo link.Once unrared, you should find the following directory tree:
raw_ovary/ ├── w5_134934.tif ├── w10_112648.tif ├── w22_090202.tif ├── w31_084030.tif ├── w40_094116.tif ├── w50_142422.tif └── w60_155112.tif
Image and data requirements
When adapting this pipeline to your own data, the following requirements should be met:
Input images must be 3D TIFF volumes.
Typical axis order is
ZYX(single channel).Expected physical resolution is approximately
(5.0, 0.867, 0.867)µm in(Z, Y, X).If your data use a different resolution, resampling to this scale is recommended for best results.
Model training
Again, BiaPy offers different options to run this training workflow depending on your level of computer expertise. Select the one that is most appropriate for you:
This training setup uses the OneDrive sample dataset (oocyte_training.zip) as training data and the Zenodo ovary volumes (raw_ovary.rar) as the test set (see Data preparation).
Download the training YAML file here:
First, download the training configuration file ovarian_reserve_training.yaml.
Next, in BiaPy’s GUI, follow the following instructions:
Step 1: Click on "Load and modify workflow" and select the ovarian_reserve_training.yaml file you just downloaded.
Step 2: Click on "OK".
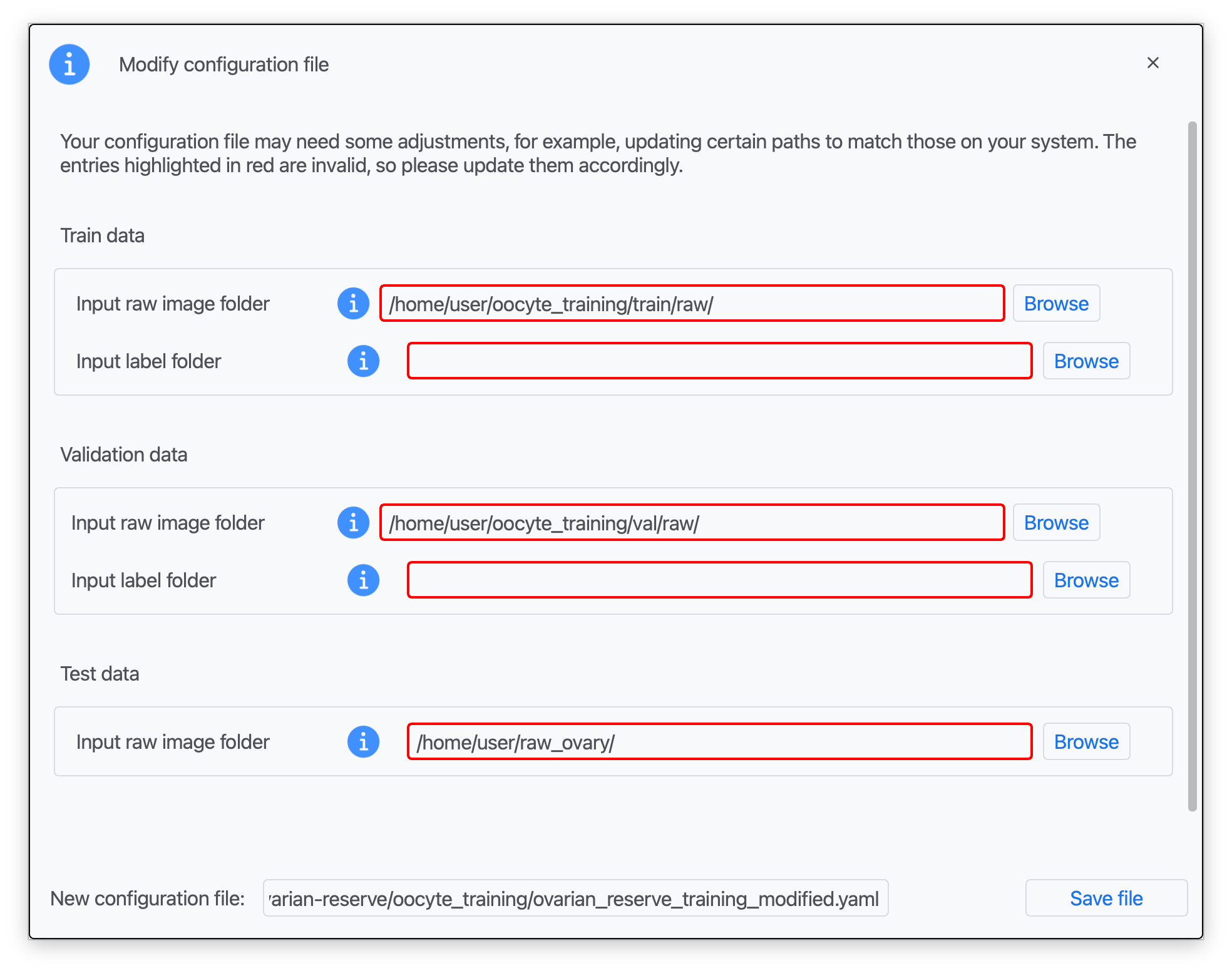
Step 3: Introduce the paths to your training raw and label data (oocyte_training/train/raw/ and oocyte_training/train/label/), validation data (oocyte_training/val/raw/ and oocyte_training/val/label/), set the test path to raw_ovary/ (Zenodo), and type a name for your modified configuration file (see Data preparation; red boxes indicate missing information).
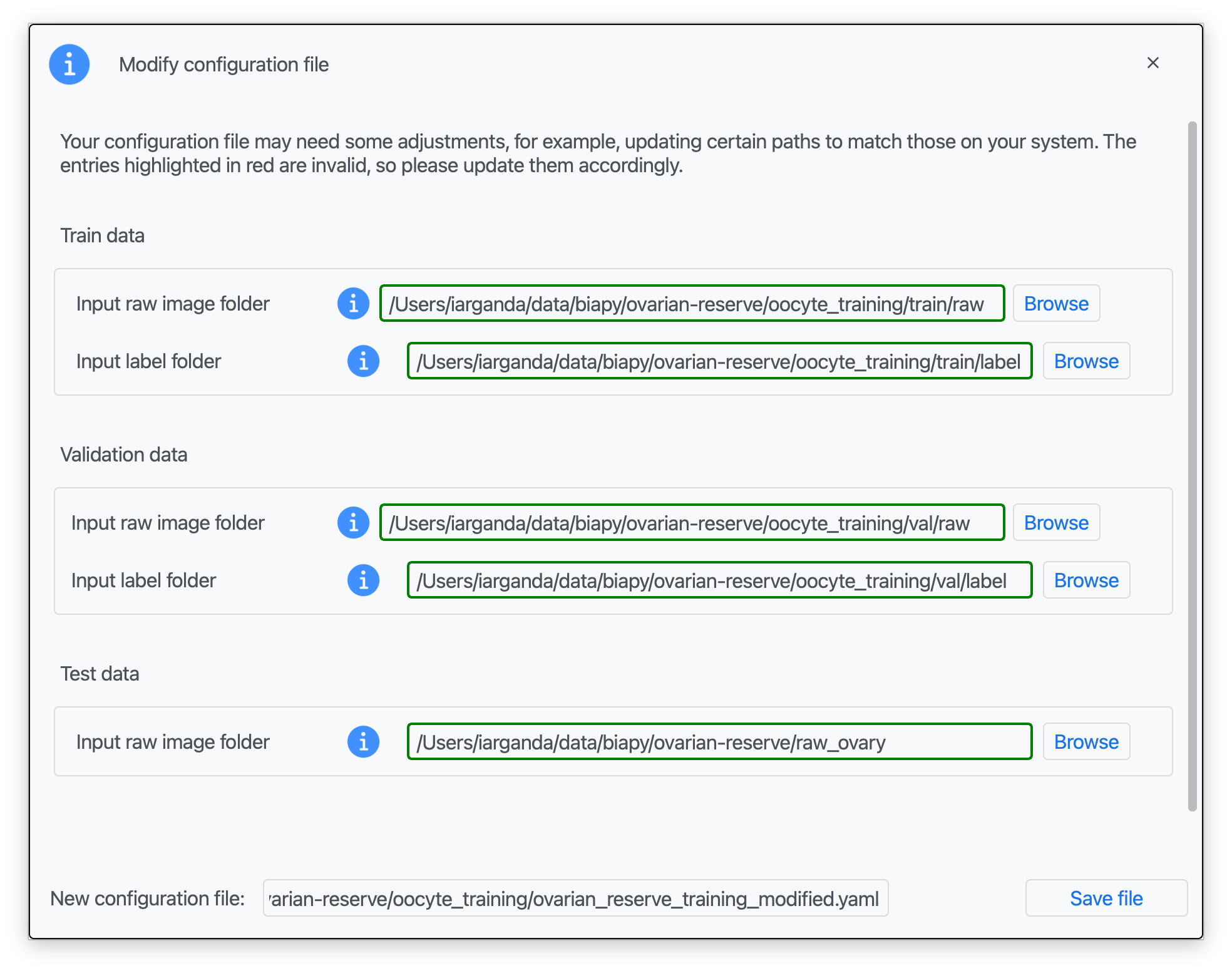
Step 4: Once that information is correctly introduced, click on "Save File".
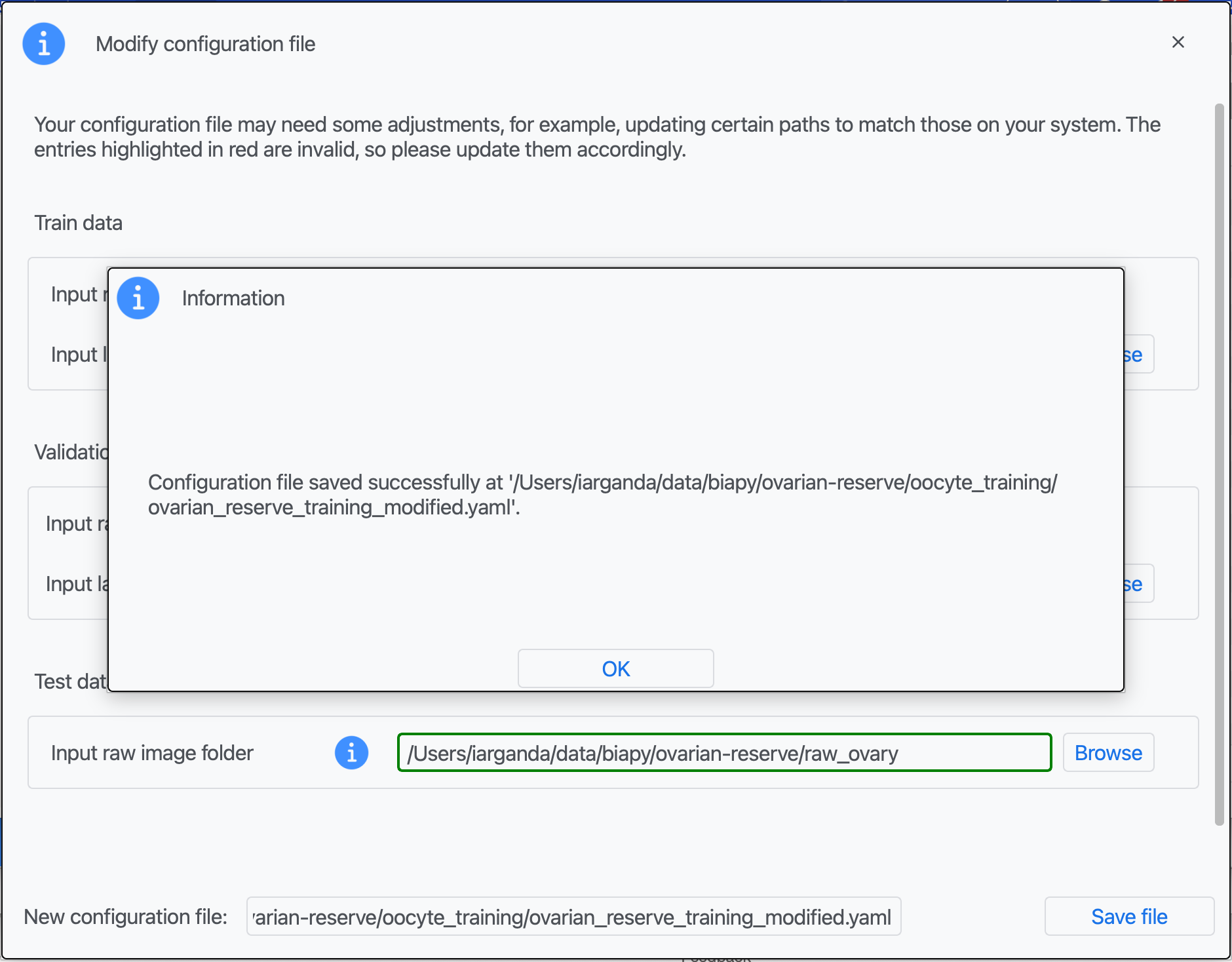
Step 5: A success window should appear. Click on "OK".
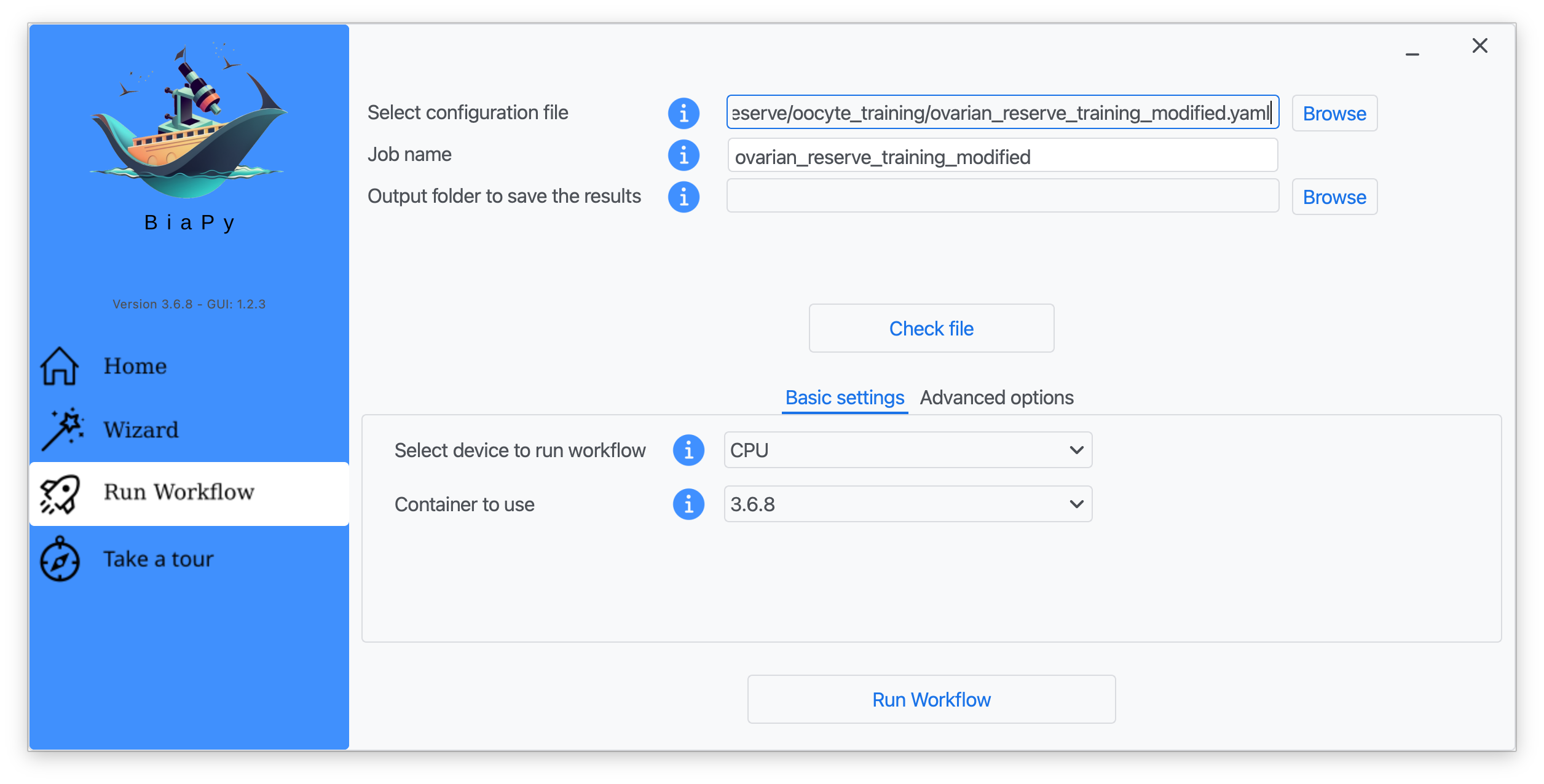
Step 6: Input the folder you wish to use to store the results of the workflow by clicking on the "Browse" button of "Output folder to save the results", and introduce or modify the proposed experiment name in the "Job name" field.
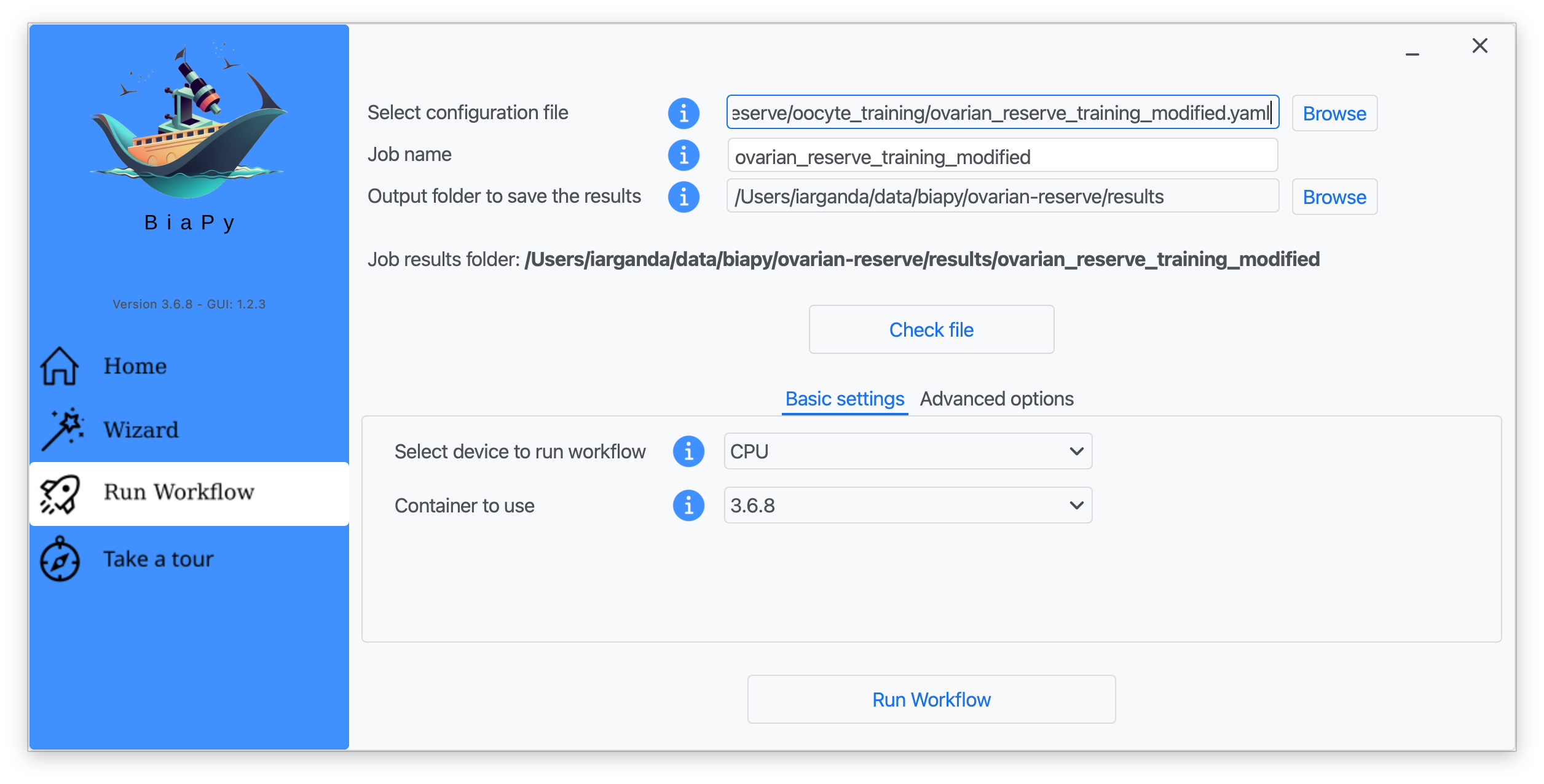
Step 7: Once filled, click on "Check File".
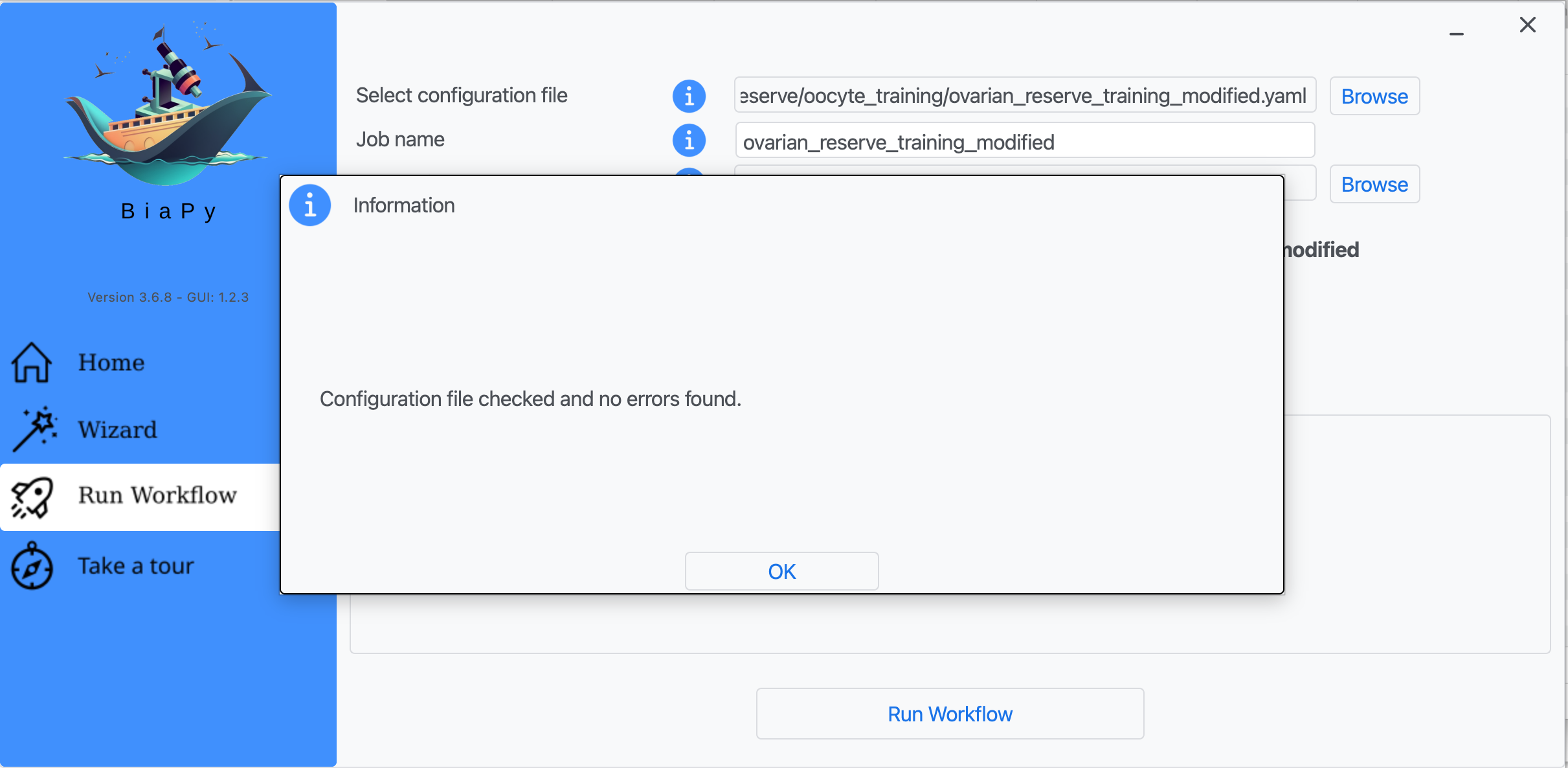
Step 8: A success window should appear. Click on "OK" and then on "Run Workflow".
Note
BiaPy’s GUI requires that all data and configuration files reside on the same machine where the GUI is being executed.
Tip
If you need additional help with the parameters of the GUI, watch BiaPy’s GUI walkthrough video.
Open the ovarian reserve training notebook in Google Colab: ![]()
This notebook is tailored to this tutorial and guides you through training in a step-by-step way, including where to place your training (oocyte_training/train/), validation (oocyte_training/val/), and test data (raw_ovary/).
Tip
If you need additional help, watch BiaPy’s Notebook walkthrough video.
Open BiaPy in Galaxy using this launch link.
Then upload your training raw/label images from oocyte_training/train/, validation raw/label images from oocyte_training/val/, upload the test volumes from raw_ovary/ (see Data preparation), select ovarian_reserve_training.yaml as configuration file, and run the job.
First, download the training configuration file ovarian_reserve_training.yaml and edit it to set the correct paths to oocyte_training/train/raw/, oocyte_training/train/label/, oocyte_training/val/raw/, oocyte_training/val/label/ and the test set from raw_ovary/ (see Data preparation).
Then, open a terminal as described in Installation and execute the following commands:
job_cfg_file=/home/user/ovarian_reserve_training.yaml
data_dir=/home/user/data
result_dir=/home/user/exp_results
job_name=my_ovarian_reserve_training
job_counter=1
gpu_number=0
docker run --rm \
--gpus "device=$gpu_number" \
--mount type=bind,source=$job_cfg_file,target=$job_cfg_file \
--mount type=bind,source=$result_dir,target=$result_dir \
--mount type=bind,source=$data_dir,target=$data_dir \
biapyx/biapy:latest-11.8 \
biapy \
--config $job_cfg_file \
--result_dir $result_dir \
--name $job_name \
--run_id $job_counter \
--gpu "$gpu_number"
Note
Note that data_dir must contain all the paths referenced by DATA.TRAIN.PATH, DATA.TRAIN.GT_PATH, DATA.VAL.PATH, DATA.VAL.GT_PATH and DATA.TEST.PATH so the container can find them.
First, download the training configuration file ovarian_reserve_training.yaml and edit it to set the correct paths to oocyte_training/train/raw/, oocyte_training/train/label/, oocyte_training/val/raw/, oocyte_training/val/label/ and the test set from raw_ovary/ (see Data preparation).
Next, run the following commands from a terminal:
job_cfg_file=/home/user/ovarian_reserve_training.yaml
result_dir=/home/user/exp_results
job_name=my_ovarian_reserve_training
job_counter=1
gpu_number=0
conda activate BiaPy_env
biapy \
--config $job_cfg_file \
--result_dir $result_dir \
--name $job_name \
--run_id $job_counter \
--gpu "$gpu_number"
Before running the command, make sure to update the following parameters:
job_cfg_file: full path to the ovarian reserve training configuration file.
result_dir: full path to the folder where results will be stored. A new subfolder will be created within this folder for each run.
job_name: a name for your experiment. Tip: avoid using hyphens (-) or spaces in the name.
job_counter: a number to identify each execution of your experiment. Start with1and increase it if you run the experiment multiple times.
If you prefer to integrate the workflow into your own Python code, you can run the same training setup through the BiaPy API once DATA.TRAIN.PATH, DATA.TRAIN.GT_PATH, DATA.VAL.PATH, DATA.VAL.GT_PATH and DATA.TEST.PATH are correctly defined in ovarian_reserve_training.yaml (see Data preparation).
from biapy import BiaPy
config_path = "/home/user/ovarian_reserve_training.yaml"
result_dir = "/home/user/exp_results"
job_name = "my_ovarian_reserve_training"
biapy = BiaPy(config_path, result_dir=result_dir, name=job_name, run_id=1, gpu="0")
biapy.run_job()
Model testing
Again, BiaPy offers different options to run this prediction workflow (also called inference) depending on your level of computer expertise. Select the one that is most appropriate for you:
Download the prediction YAML file here:
Note
If you do not have a checkpoint yet, you can download the pretrained model from this SharePoint link. You can also generate your own checkpoint by following Model training.
Note
The test data are TIFF volumes, and the provided YAML is already configured for TIFF input axis order. Since these volumes are usually large, they are processed internally in chunks as Zarr for efficiency, while final predictions can still be exported as TIFF (controlled with the TEST.BY_CHUNKS.SAVE_OUT_TIF parameter).
First, download the prediction configuration file ovarian_reserve_inference.yaml and prepare a pretrained .pth model checkpoint (either your own from Model training or the provided pretrained model from the SharePoint link above).
Next, in BiaPy’s GUI, follow the following instructions:
Step 1: Click on "Load and modify workflow" and select the ovarian_reserve_inference.yaml file you just downloaded.
Step 2: Click on "OK".
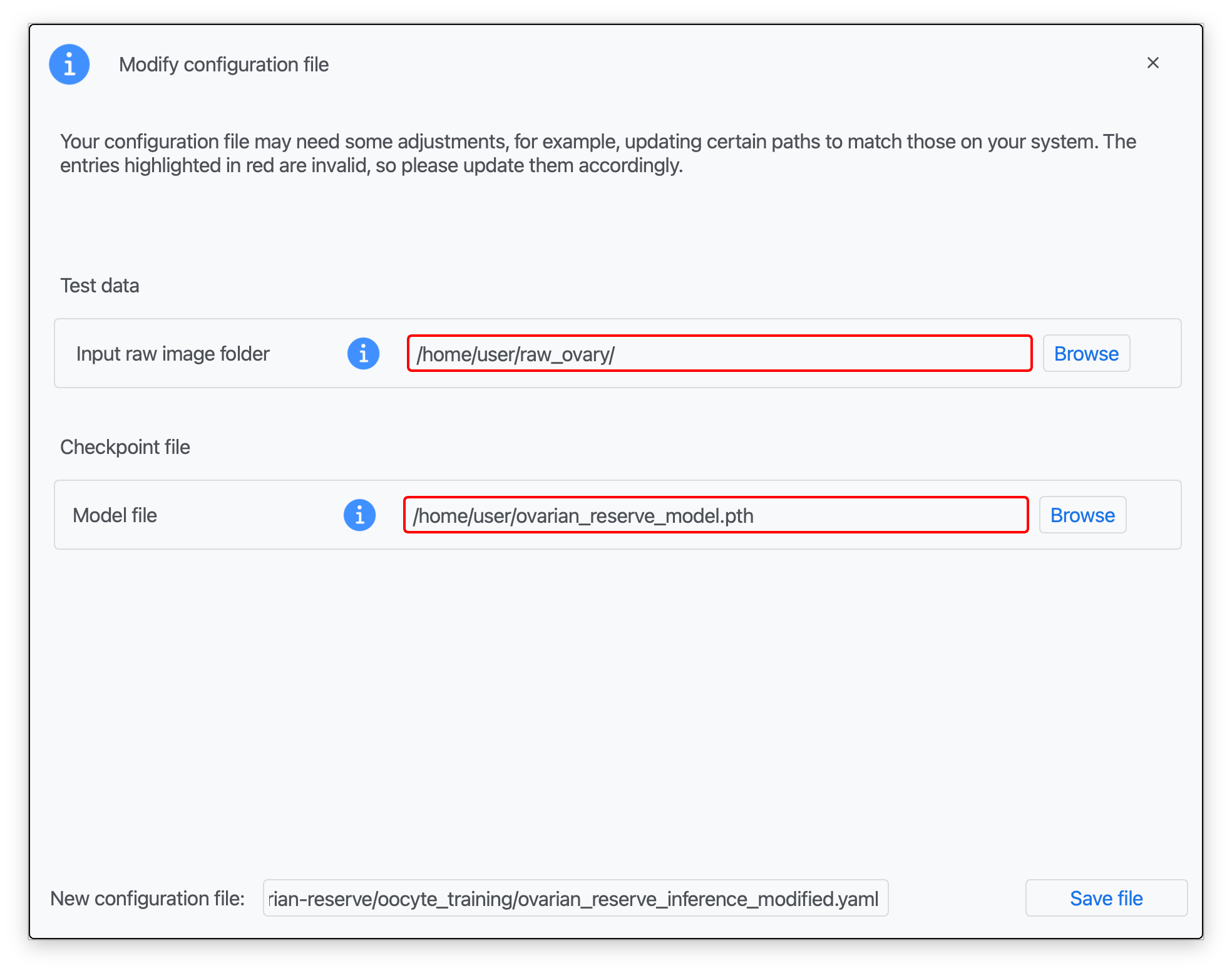
Step 3: Introduce the corresponding paths to your test data (raw images), a pretrained .pth model file, and a name for your modified configuration file (see Data preparation; red boxes indicate missing information).
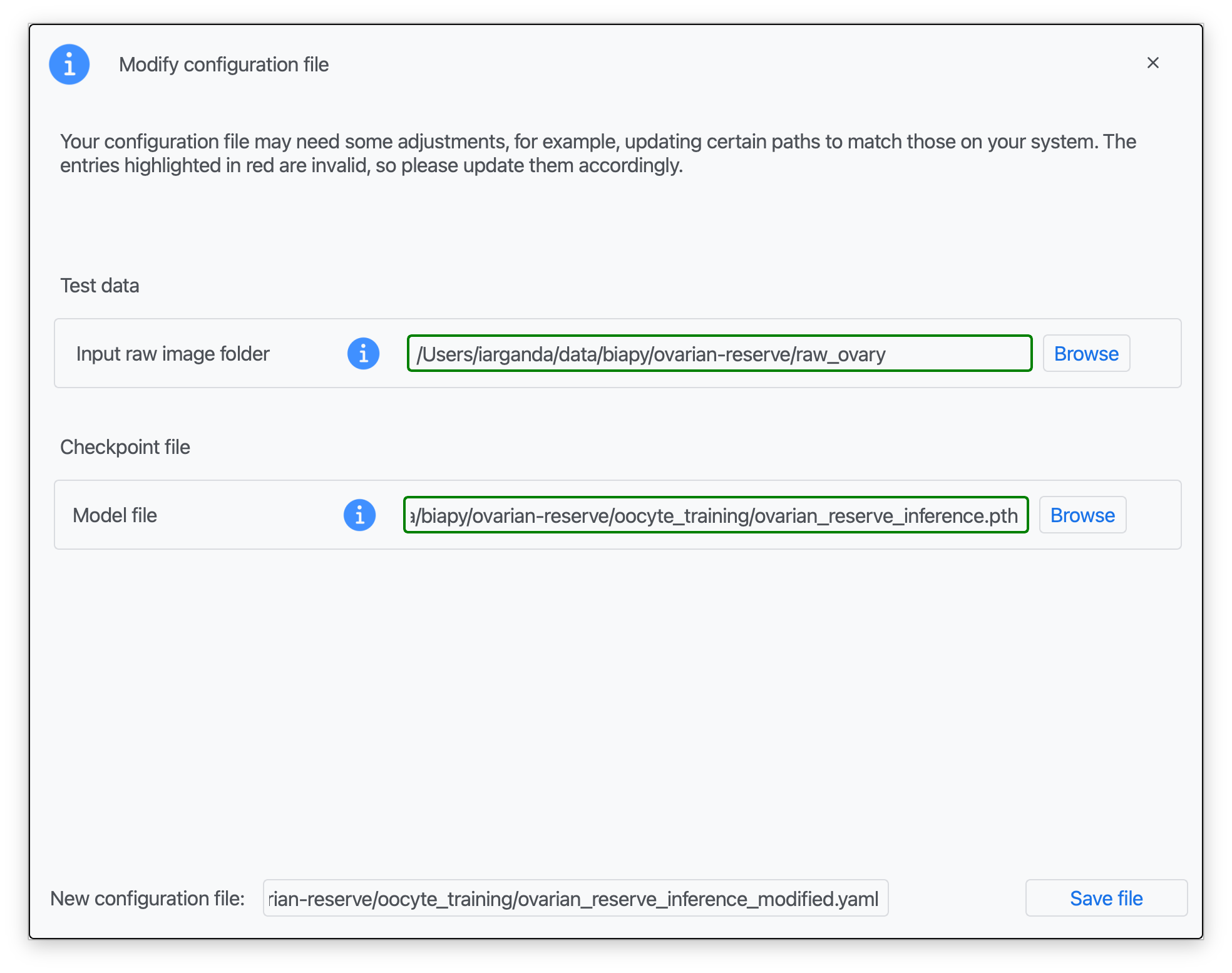
Step 4: Once that information is correctly introduced, click on "Save File".
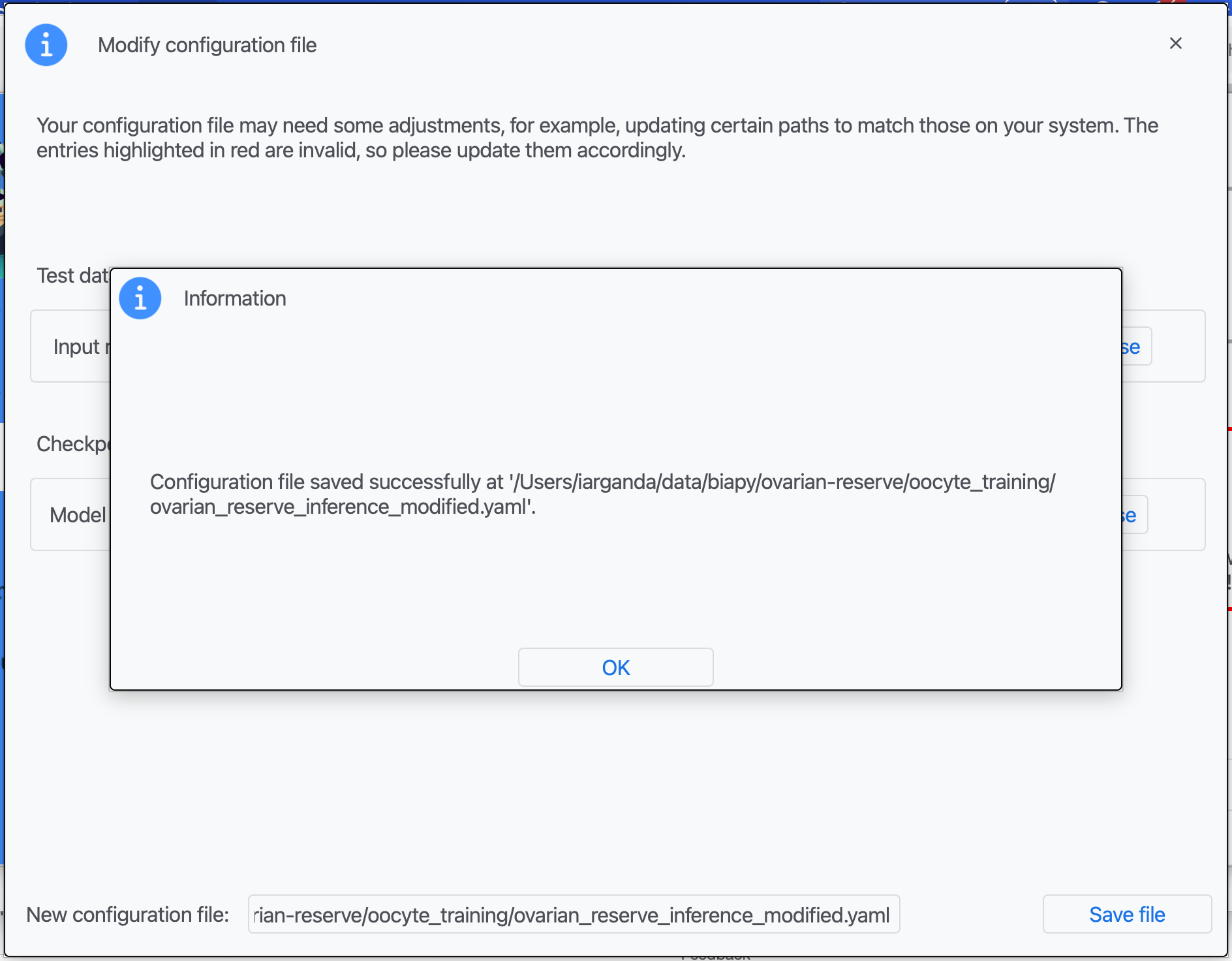
Step 5: A success window should appear. Click on "OK".
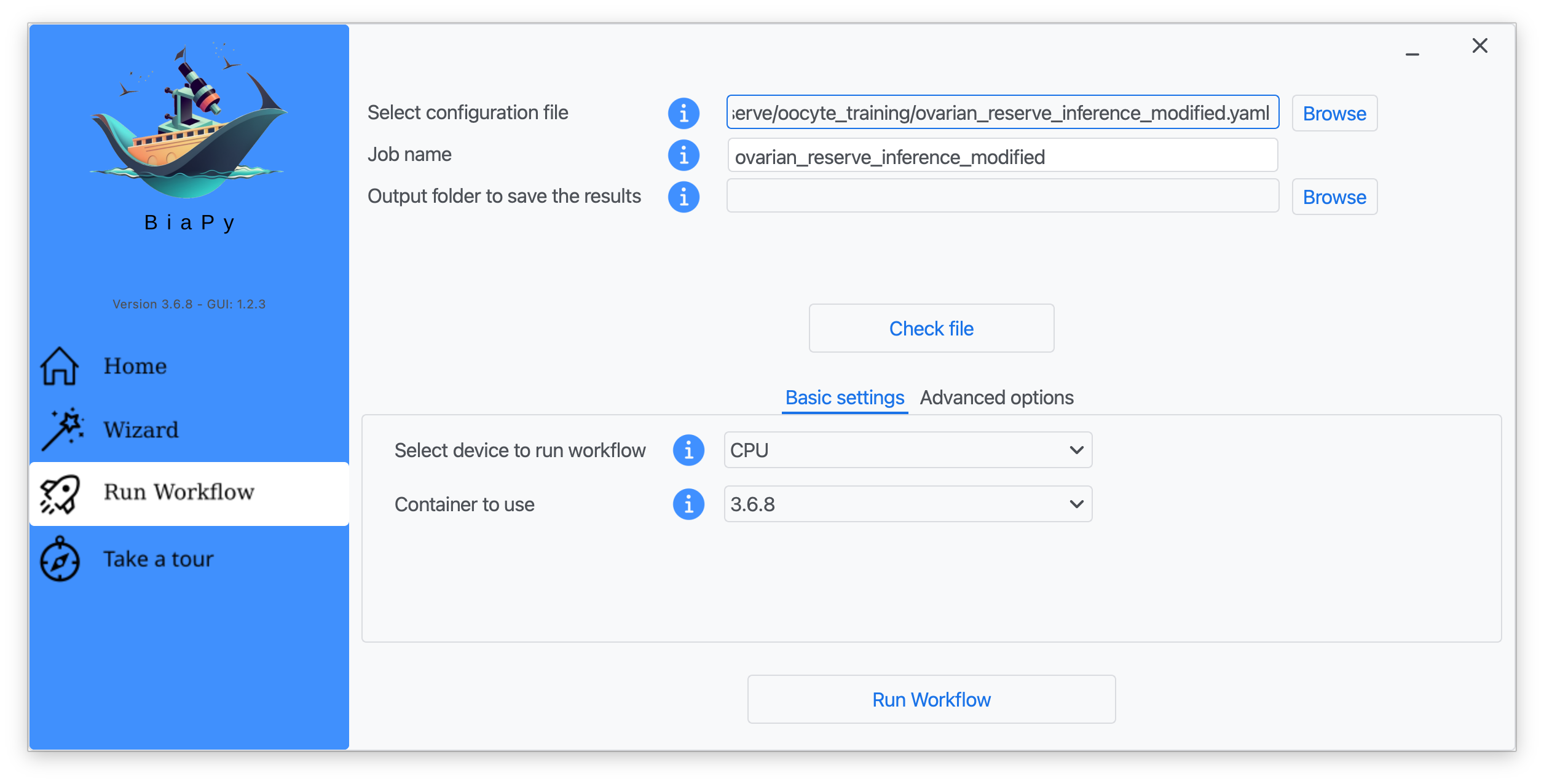
Step 6: Input the folder you wish to use to store the results of the workflow by clicking on the "Browse" button of "Output folder to save the results".
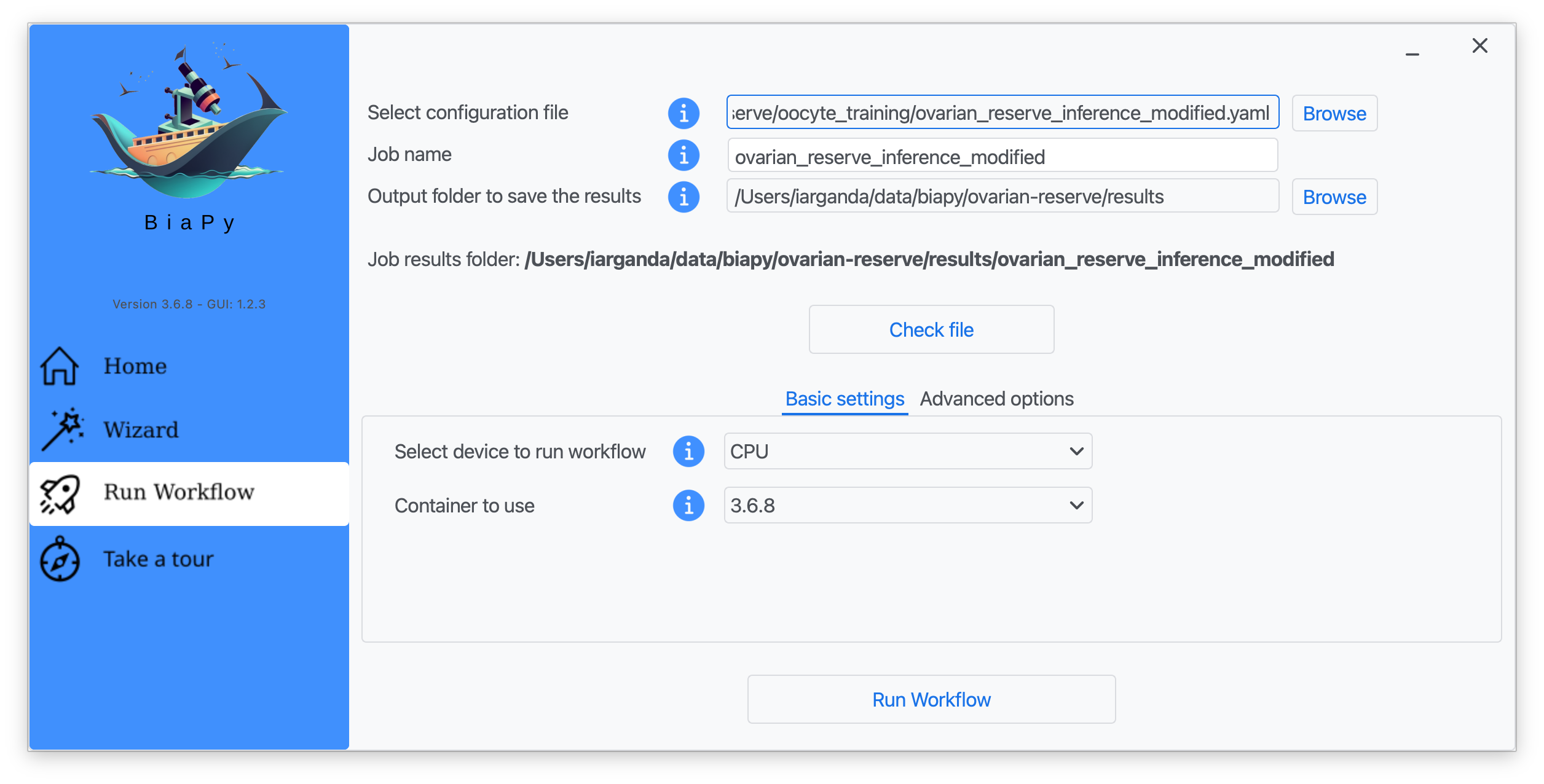
Step 7: Once filled, click on "Check File".
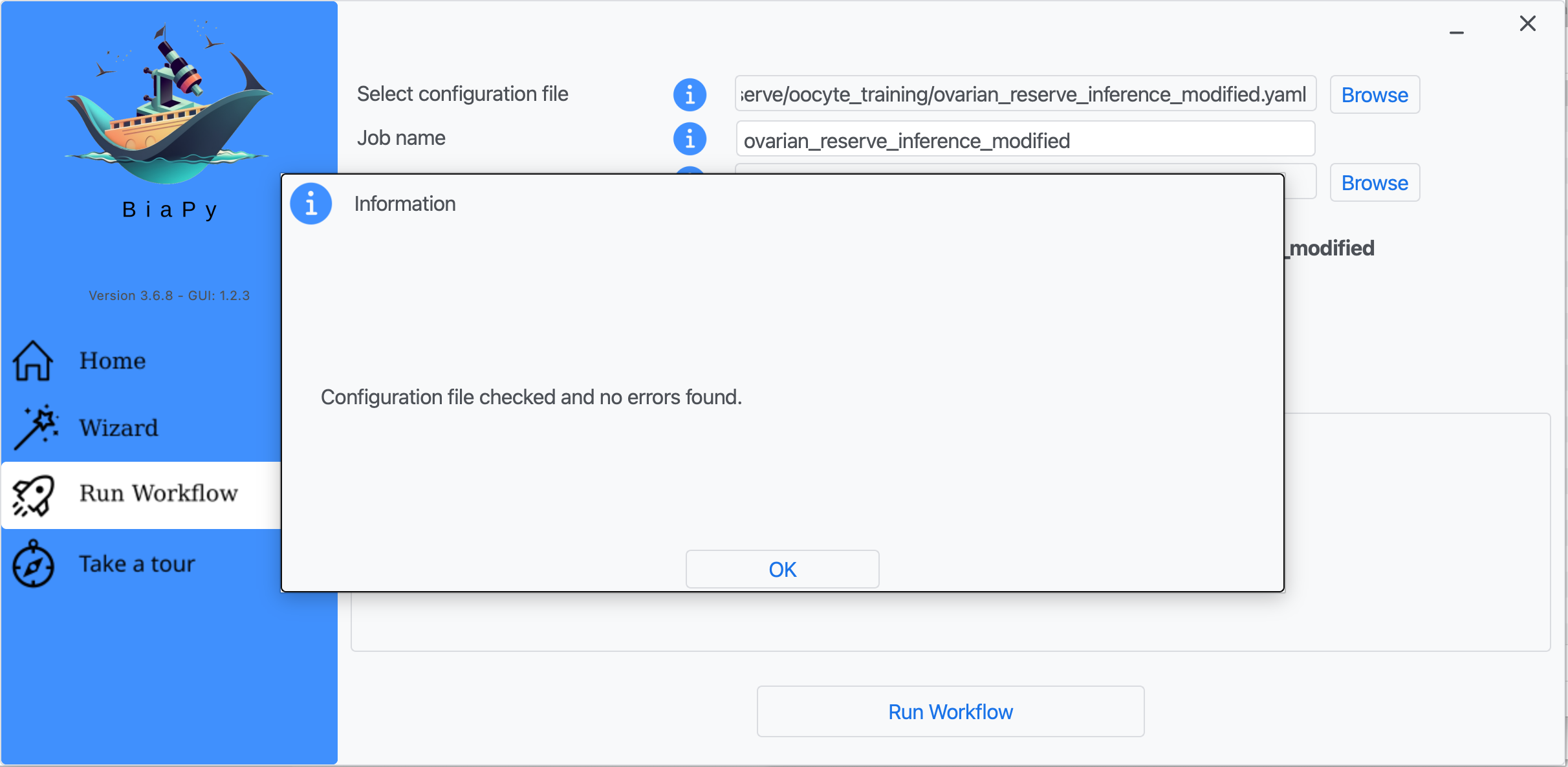
Step 8: A success window should appear. Click on "OK" and then on "Run Workflow".
Note
BiaPy’s GUI requires that all data and configuration files reside on the same machine where the GUI is being executed.
Tip
If you need additional help with the parameters of the GUI, watch BiaPy’s GUI walkthrough video.
Open the ovarian reserve inference notebook in Google Colab: ![]()
This notebook is focused on prediction (inference). You will load your ovary images, provide a model checkpoint (for example, the pretrained model linked above), and generate the oocyte instance segmentations.
Tip
If you need additional help, watch BiaPy’s Notebook walkthrough video.
Open BiaPy in Galaxy using this launch link.
Then upload your TIFF images from raw_ovary/ (see Data preparation), upload ovarian_reserve_inference.yaml, provide the checkpoint file, and run the job to download the predictions.
First, download the prediction configuration file ovarian_reserve_inference.yaml and prepare a pretrained checkpoint. Next edit the configuration file to set the correct path to your test data folder from raw_ovary/ (DATA.TEST.PATH) and the pretrained model (PATHS.CHECKPOINT_FILE).
Then, open a terminal as described in Installation and execute the following commands:
job_cfg_file=/home/user/ovarian_reserve_inference.yaml
data_dir=/home/user/raw_ovary
result_dir=/home/user/exp_results
job_name=my_ovarian_reserve_test
job_counter=1
gpu_number=0
docker run --rm \
--gpus "device=$gpu_number" \
--mount type=bind,source=$job_cfg_file,target=$job_cfg_file \
--mount type=bind,source=$result_dir,target=$result_dir \
--mount type=bind,source=$data_dir,target=$data_dir \
biapyx/biapy:latest-11.8 \
biapy \
--config $job_cfg_file \
--result_dir $result_dir \
--name $job_name \
--run_id $job_counter \
--gpu "$gpu_number"
Note
Note that data_dir must contain the folder pointed to by DATA.TEST.PATH so the container can find it. For instance, in this example DATA.TEST.PATH could be /home/user/raw_ovary.
First, download the prediction configuration file ovarian_reserve_inference.yaml and prepare a pretrained checkpoint. Next edit the configuration file to set the correct path to your test data folder from raw_ovary/ (DATA.TEST.PATH) and the pretrained model (PATHS.CHECKPOINT_FILE; see Data preparation).
Next, run the following commands from a terminal:
job_cfg_file=/home/user/ovarian_reserve_inference.yaml
result_dir=/home/user/exp_results
job_name=my_ovarian_reserve_test
job_counter=1
gpu_number=0
conda activate BiaPy_env
biapy \
--config $job_cfg_file \
--result_dir $result_dir \
--name $job_name \
--run_id $job_counter \
--gpu "$gpu_number"
Before running the command, make sure to update the following parameters:
job_cfg_file: full path to the ovarian reserve prediction configuration file.
result_dir: full path to the folder where results will be stored. A new subfolder will be created within this folder for each run.
job_name: a name for your experiment. Tip: avoid using hyphens (-) or spaces in the name.
job_counter: a number to identify each execution of your experiment. Start with1and increase it if you run the experiment multiple times.
If you prefer to integrate the workflow into your own Python code, you can run the same prediction setup through the BiaPy API once DATA.TEST.PATH and PATHS.CHECKPOINT_FILE are correctly defined in ovarian_reserve_inference.yaml (see Data preparation).
from biapy import BiaPy
config_path = "/home/user/ovarian_reserve_inference.yaml"
result_dir = "/home/user/exp_results"
job_name = "my_ovarian_reserve_test"
biapy = BiaPy(config_path, result_dir=result_dir, name=job_name, run_id=1, gpu="0")
biapy.run_job()
Results
Training results. Assuming you named your training job my_ovarian_reserve_training, the results should be stored in the folder defined in result_dir, with a structure similar to this:
my_ovarian_reserve_training/
├── config_files/
│ └── ovarian_reserve_training.yaml
├── checkpoints/
│ └── my_ovarian_reserve_training_1-checkpoint-best.pth
├── train_logs/
│ └── my_ovarian_reserve_training_1_log_....txt
└── results/
└── my_ovarian_reserve_training_1/
├── aug/
├── charts/
└── tensorboard/
Where:
config_files: directory where YAML files used in the experiment are stored.checkpoints: directory where model weights are stored.train_logs: directory where training logs are stored.results: directory where generated checks and outputs are stored, with one subfolder per run.
Testing results. Assuming you named your testing job my_ovarian_reserve_test, the results should be stored in the folder defined in result_dir, with a structure similar to this:
my_ovarian_reserve_test/
├── config_files/
│ └── ovarian_reserve_inference.yaml
└── results/
└── my_ovarian_reserve_test_1/
├── per_image/
├── per_image_instances/
├── per_image_post_processing/
└── instance_associations/
Where:
config_files: directory where YAML files used in the experiment are stored.results: directory where generated checks and outputs are stored, with one subfolder per run.per_image: reconstructed output channel predictions.per_image_instances: final instance segmentations.per_image_post_processing: instance predictions after post-processing.instance_associations: optional CSV/TIFF summaries of instance matching against ground truth (if available).
Pre-trained models in the BioImage Model Zoo
The model produced during this study is publicly available in the BioImage Model Zoo (BMZ) — a community-driven repository of ready-to-use deep learning models for bioimage analysis. It’s name is plucky-leopard and can be found in this link: https://bioimage.io/#/artifacts/plucky-leopard.

The oocyte segmentation model (plucky-leopard).
Note
The model can be downloaded and run directly through BiaPy or any other BMZ-compatible tool. Find more information about how to use BMZ models in BiaPy in BioImage Model Zoo documentation section.
Post-analysis scripts
After segmentation, you can run the analysis scripts from the Boke-Lab ovarian_reserve repository:
oocyte density: quantifies oocytes per volume.
radial quantification: measures the radial spatial distribution of oocytes.
These scripts reproduce the quantitative analyses described in [2].
Citation
Please note that this tutorial is based on a publication. If you use it successfully for your research, please cite our work:
3D Mapping of Intact Ovaries Reveals the Aging Dynamics of the Ovarian Reserve
Arturo D'Angelo, Daniel Franco-Barranco, Marco Musy, James Sharpe, Ignacio Arganda-Carreras,
Elvan Böke
bioRxiv 2025.11.07.686728; doi: https://doi.org/10.1101/2025.11.07.686728